General Model
Elemental Property
Elemental Property provides a comprehensive library that houses the latest and updated physical, chemical, atomic, and electronic properties of elements, including zunger-pseudopotential-radii, van-der-waals-radii,teatum-metallic-radii,mendeleev-number,crystal-structure,valence-electron-number, pauling-electronegativity, martynov-batsanov-electronegativity, etc., enabling researchers and professionals to access accurate and up-to-date data for their scientific endeavors.
Get Started >
Miedema Calculator
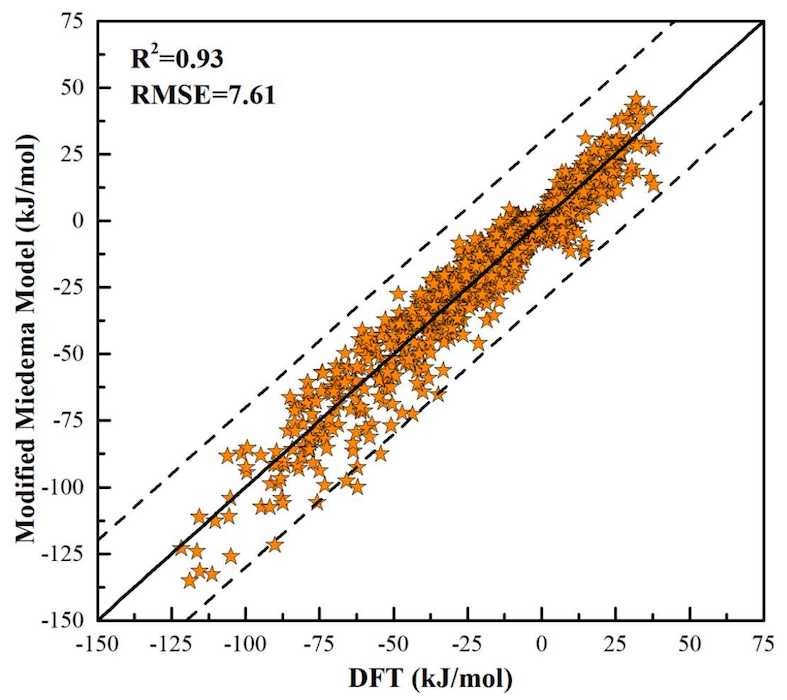
Miedema Calculator is a thermodynamic software for predicting formation enthalpies of various alloys within framework of Miedema’s Theory. The name of the software comes from the name of the famous scientist Andries Miedema. In memory of his great contribution to the empirical thermodynamical model of “Cohesion in metals”, we name this software as “Miedema Calculator”. Miedema Calculator can perform various functions such as calculation, database selection, parameter query, etc. The webpage version of Miedema Calculator provides the calculation of the standard formation enthalpy of binary intermetallic compounds based on the ML optimized parameters.
Get Started >Ternary Diagram
Ternary Diagram is a sophisticated thermodynamic software that employs the Miedema calculator to predict the formation enthalpies of ternary alloys. Based on Miedema's groundbreaking theory of 'Cohesion in Metals,' this software accurately calculates the energetics of alloy formation by considering the physical and chemical properties of the constituent elements. It provides valuable insights into the thermodynamic behavior of ternary alloys, facilitating the design of novel alloy compositions with desired properties.
Get Started >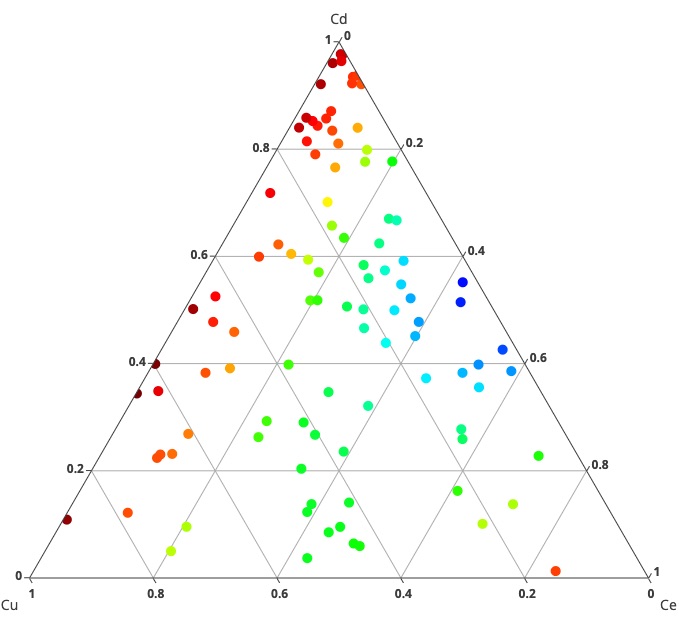
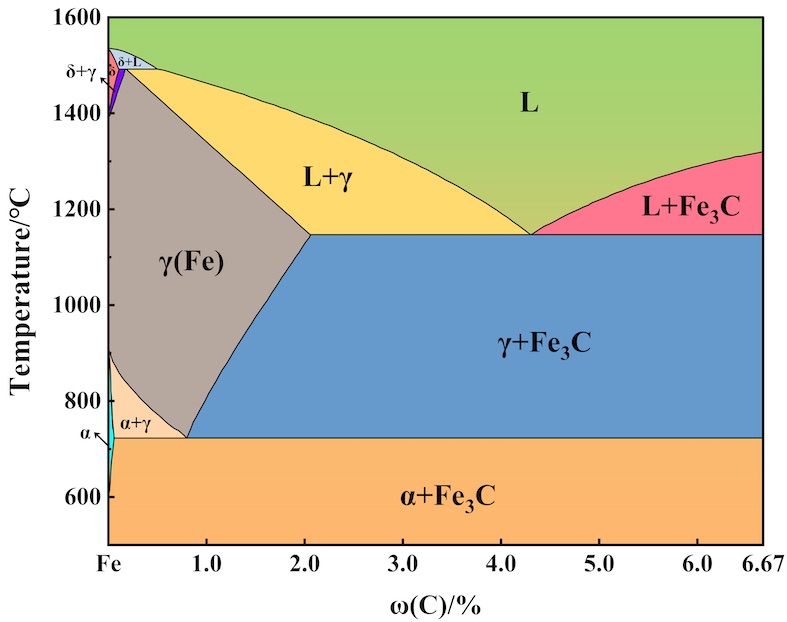
Alloy Phase Diagram
Phase Diagram, also known as the phase equilibrium diagram, is a crucial tool in the fields of physics, chemistry, and materials science. It serves to illustrate the relationship between the composition of a system and its thermodynamic parameters, such as temperature and pressure. The diagram is a visual representation of the various phases that an alloy system can exist in, as well as the conditions under which phase transitions occur. For instance, in a binary alloy system, the phase diagram depicts the stable and unstable regions of different alloy phases as a function of temperature and composition.
Get Started >Pourbaix Calculator
Pourbaix Calculator is an electrochemical thermodynamic calculation software that is used to calculate the Pourbaix diagram, i.e. an electrochemical phase diagram relative to the solution pH and electrode potential E (using a standard hydrogen electrode as a reference). At present, the Pourbaix diagrams of metals, binary alloys, and two-dimensional materials (MXenes) can all be calculated when the solution contains specific inorganic ions, such as Cl-, CO32-. Pourbaix Calculator supports functions such as database selection and user input, system parameter settings, calculation of Pourbaix diagrams, and output of calculation results.
Get Started >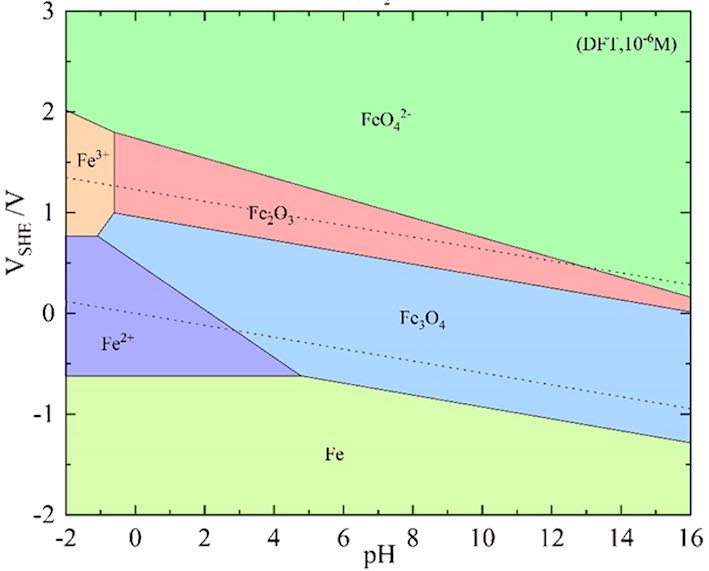
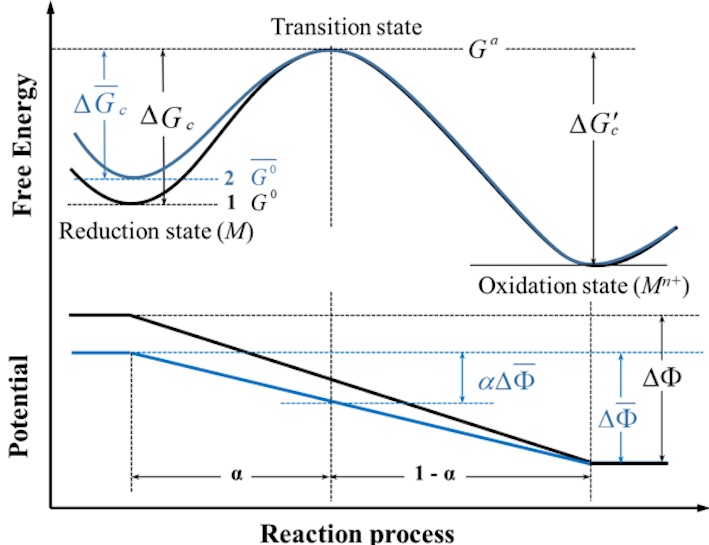
EPCK Analyzer
EPCK Analyzer is a user interface program for electrochemical polarization and corrosion kinetics of metals and alloys based on their surface properties, such as surface energy and working function. EPCK includes multiple functional modules: Calculate the equilibrium exchange current density and equilibrium potential of metals and alloys relative to standard hydrogen electrodes (SHE); Calculate the polarization curve based on the surface energy and work function of the characteristic crystal planes of various metals; Calculate the polarization curve based on the surface energy and work function of various doped alloys; Calculate the impact of different corrosion conditions on the corrosion resistance of metal surfaces; Calculate the effect of mechanochemical coupling on the dynamic behavior of metal surface corrosion.
Get Started >Symmetry Analyzer
Symmetry Analyzer is user interface program to analyze the crystal symmetry of a given materials, based on the spglib program as implemented in VASPMATE. Symmetry is the most fundamental characteristic of crystal structures. Using this analyzer will output basic symmetry information of the crystal, such as space group, point group, crystal system, etc. This analyzer is primarily used to analyze the symmetry information of the uploaded POSCAR file.
Get Started >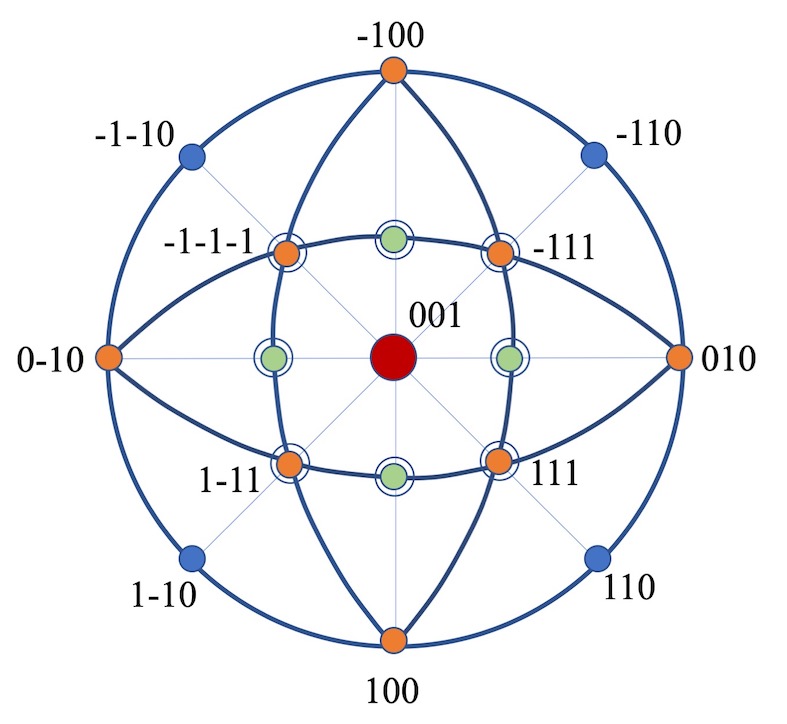
KPATH Generator
KPATH Generator is a user interface program for the generation of special K-Path high-symmetry point paths for a crystal structure that are completely consistent with SeeK-Path for 24 variant structures of the seven crystal systems. In the process of calculating the energy band structure, it is often necessary to convert the current unit cell into a normalized primitive cell and generate a high-symmetry K-point path in the Brillouin zone. This generator is mainly used to generate the normalized primitive cell form of the current unit cell, and a high-symmetry K-point path for energy band structure drawing.
Get Started >Enthalpy Predictor
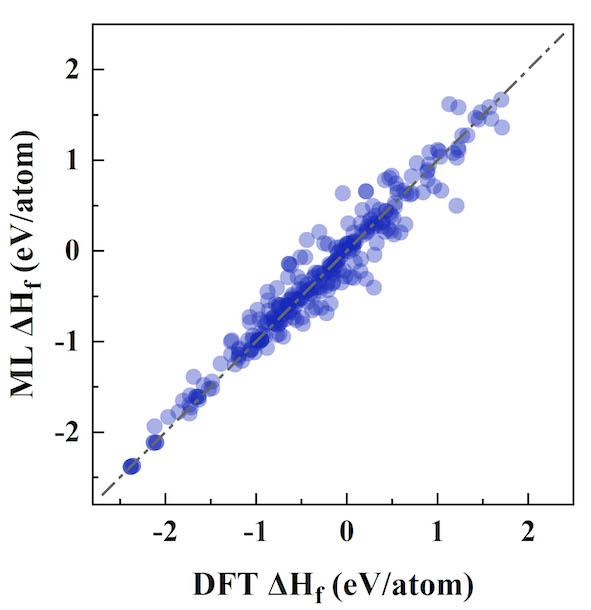
Enthalpy Predictor is a thermodynamic software for predicting formation enthalpies of various alloys by means of classification enhanced surrogate model according to Miedema's theory of "Cohesion in metals". Enthalpy Predictor can perform calculation of the standard formation enthalpy of binary intermetallic compounds with high precise by simply providing a POSCAR. The feature importance analysis highlight the sufficiency of the present ML model to characterize high-dimensional space by adopting a physics-informed classification strategy.
Get Started >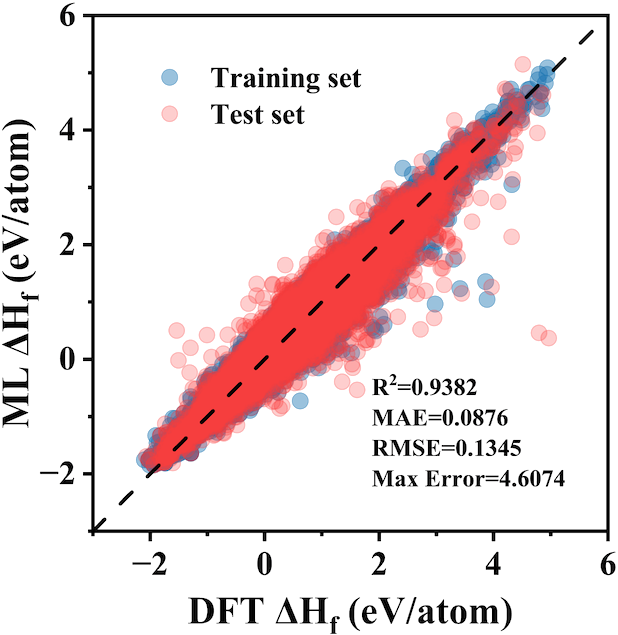

Virtural Diffraction Analysis
Virtural Diffraction Analysis is derived from the robust and convenient command-line program, termed as AAVDP: An automatic Analyzer for Virtural Diffraction Pattern of crystalline and noncrystalline solids, which has implemented the various virtual simulation of diffraction patterns, including the x-ray diffraction (XRD) pattern, the neutron diffraction (NED) pattern, the selected-area kinematic electron diffraction (KED) pattern, the kinematic kikuchi diffraction (KKD) pattern, and the dynamic kikuchi diffraction (DKD) pattern, as well as the analyses of the radial distribution function (RDF), and the static structure factor (SSF).
Get Started >Special Model
Alloy Immiscibility Map
The classifcation of miscible and immiscible systems of binary alloys plays a critical role in the design of multicomponent alloys. By mining data from hundreds of experimental phase diagrams, and thousands of thermodynamic data sets from experiments and high-throughput frst-principles (HTFP) calculations, a comprehensive classifcation of alloying behavior for 813 binary alloy systems consisting of transition and lanthanide metals was performed. Among several physics-based descriptors, the slightly modifed Pettifor chemical scale provides a unique two-dimensional map that divides the miscible and immiscible systems into distinctly clustered regions. Based on an artifcial neural network algorithm and elemental similarity, the miscibility of the unknown systems is further predicted and a complete miscibility map is thus obtained. Impressively, the classifcation by the miscibility map yields a robust validation on the capability of the well-known Miedema’s theory (95% agreement) and shows good agreement with the HTFP method (90% agreement).
Get Started >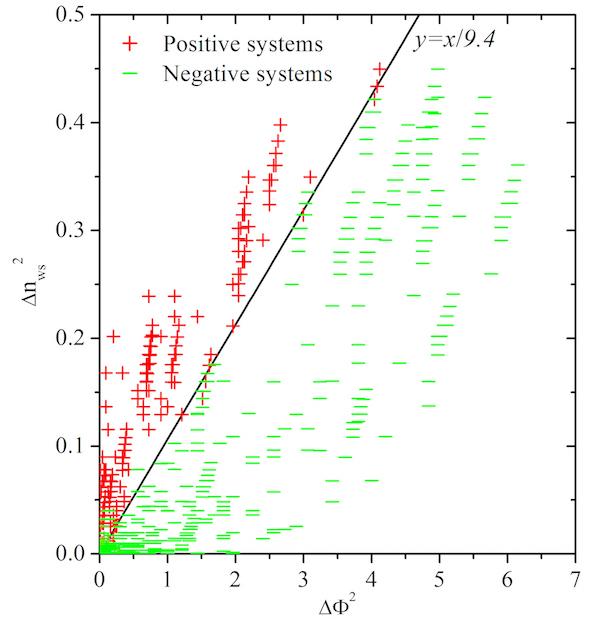
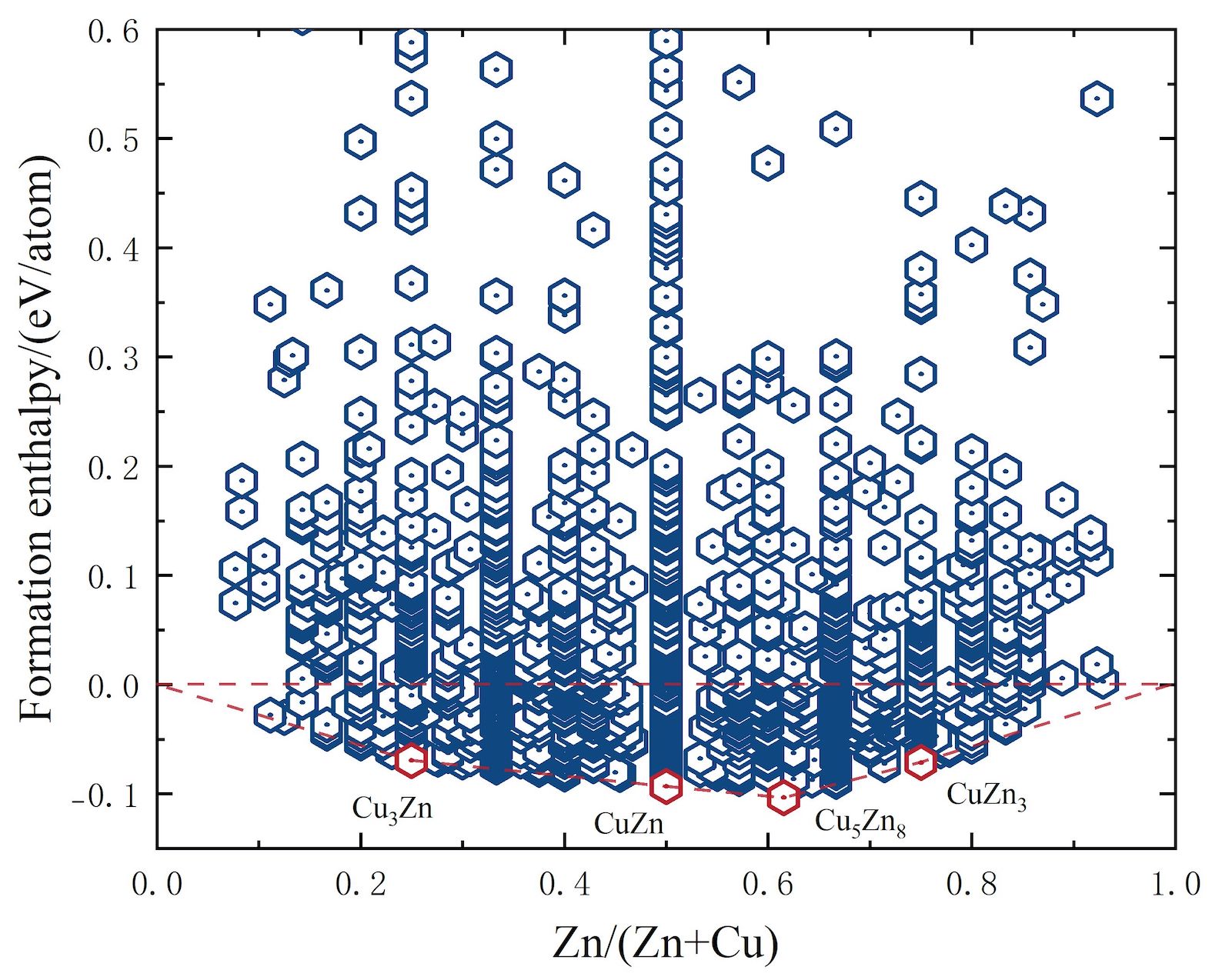
Convexhull Analyzer
High-throughput (HT) computations is a fundamental approach in data-driven paradigms owing to their efficiency in data creation. By means of HT first principles method and combinational screeening scheme, the formation enthalpies of a series of binary systems are comprehensively studied, and then a most thermodynamically stable (or experimentally feasible) ones are screened by constructing the well-known convex hull diagram, which shows consistency with the available experiments.
Get Started >Electronic Band Structure
Electronic band structure refers to the energy states of electrons in a solid material, specifically within the crystal lattice of a metal, semiconductor, or insulator. It is a fundamental concept in condensed matter physics and solid-state physics that describes how electrons behave in periodic structures. Understanding the electronic band structure is crucial for predicting and explaining various material properties such as conductivity, optical response, and magnetic behavior..
Get Started >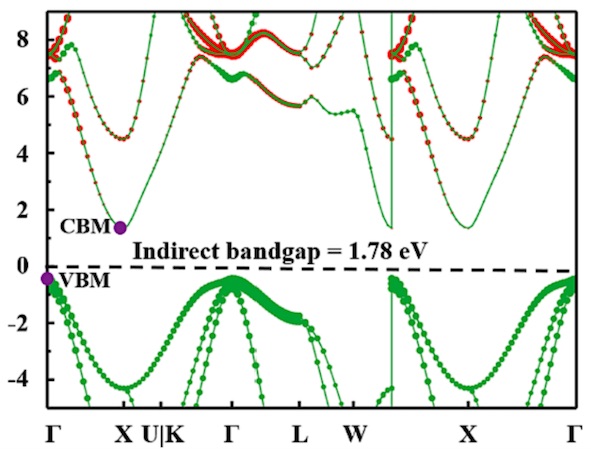
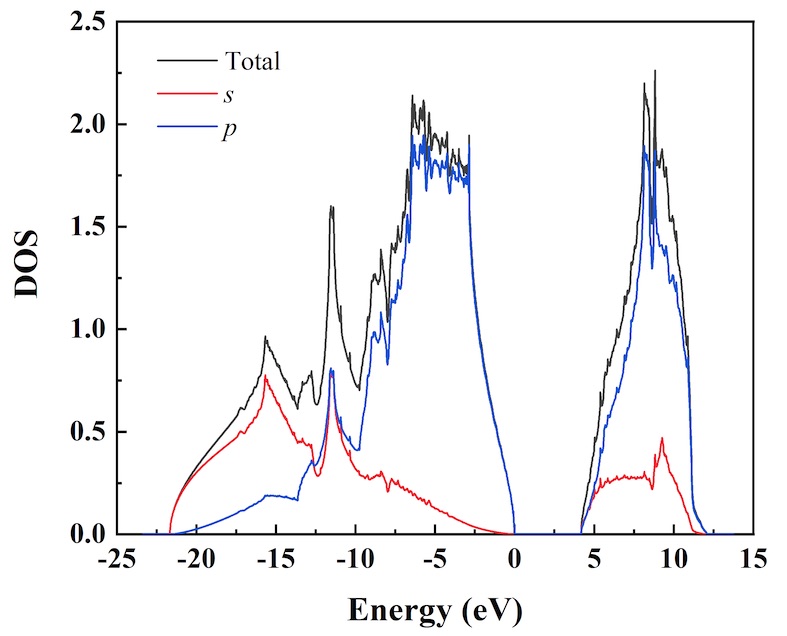
Density of state
Density of states (DoS) refers to the number of quantum states available to a system within a given range of energy. It is a fundamental concept in statistical physics and quantum mechanics that plays a crucial role in understanding the thermodynamic properties of matter. In the context of condensed matter physics, the density of states describes how the energy levels of electrons, phonons, or other particles are distributed in a solid material.
Get Started >Elasticity Anisotropy
Elasticity Anisotropy also known as elastic anisotropy, is a physical property that refers to the variation in elastic properties of a material with respect to direction. A material's elastic moduli (e.g., Young's modulus, shear modulus, and bulk modulus) vary depending on the direction of applied stress or deformation. It is a fundamental concept in various disciplines such as physics, materials science, and engineering.
Get Started >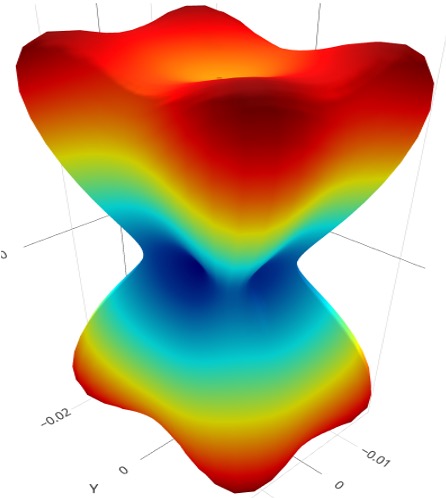
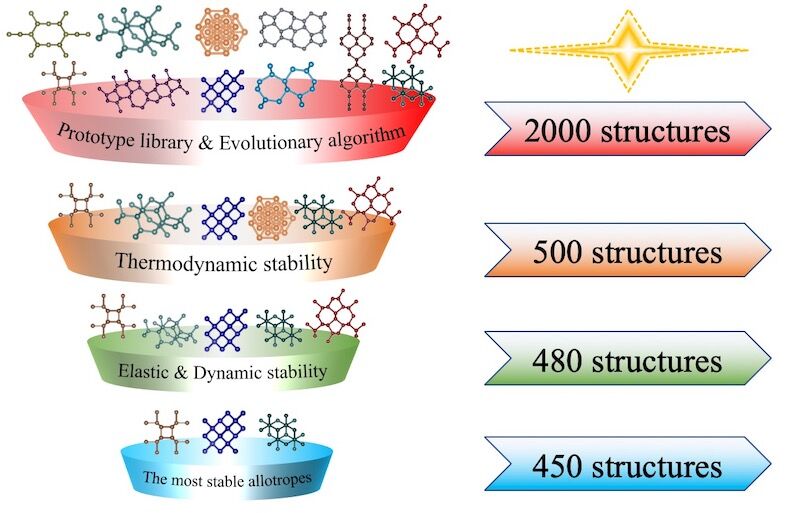
Property Predictor for carbon allotropes
High-throughput (HT) computations and machine learning (ML) modelling are two critical approaches in data-driven paradigms due to their efficiency in data creation and model construction. By combining both approaches, a high efficient surrogate model is proposed for the prediction of the thermodynamic stability and mechanical properties of carbon allotropes with high precision, providing a pathway for the quick screen the novel superhard carbon materials.
Get Started >Enthalpy predictor for superhard BN
High-throughput (HT) computations and machine learning (ML) algorithms are two fundamental approaches in data-driven paradigms to predict various properties of solids due to their efficiency in data creation and model construction, which however are usually used individually and lack generalization and flexibility. Recently, a scheme is proposed to combine HT computations for the efficient creation of consistent databases and ML algorithms for the fast construction of surrogate models in order to screen the properties of superhard BN compounds with high precision through four descriptors.
Get Started >